Genetic variation of common walnut (Juglans regia) in Piedmont, Northwestern Italy
Forest@ - Journal of Silviculture and Forest Ecology, Volume 4, Pages 386-394 (2007)
doi: https://doi.org/10.3832/efor0483-0040386
Published: Dec 20, 2007 - Copyright © 2007 SISEF
Research Articles
Guest Editors: RI.SELV.ITALIA - MiPAF Project
« Shared Research Program on Silviculture in Italy »
Collection/Special Issue: Massimo Bianchi
Abstract
The European or common walnut is a large tree prized as a multipurpose species: it provides valuable timber and produces a high-quality edible nut. The diffusion of the species in Italy has been largely influenced by the human activity, mainly through germplasm movement, selection of genotypes most suited for wood or fruit production and adaptation induced on fruit crop reproductive materials. As a consequence, genetic variability has been reduced, so that programs aimed at its preservation appear of the utmost importance. 104 walnut plants growing in Piedmont, northwestern Italy, were investigated through genetic variation scored at RAPD loci, yielded by PCR amplification of 10 decamer primers. Among the 101 studied loci, only 53 were polymorphic, showing a low level of genetic variation within the studied material. Genetic differentiation was estimated both at individual and geographical area level. Only in few cases trees growing in the same area showed to be genetically similar, while the differentiation between areas accounted for about 10% of the total variation, according to AMOVA. No significant correlation was found between genetic and geographic distances. The results of the study showed that also in Piedmont (such as it was already demonstrated in other parts of Italy) the distribution of common walnut is a direct consequence of the human activity. The selection of individual trees, to be used as basic materials for seed supply, should therefore be based mainly on phenotypic traits, rather than ecological features of the location: in species characterized by artificial diffusion, the adoption of Region of Provenance has a scarce significance and prominence should be given to the phenotype selection.
Keywords
Genetic variation, Molecular markers, Biodiversity, Conservation, Walnut
Introduzione
La struttura genetica delle popolazioni è il risultato di un complesso processo evolutivo, al quale concorrono numerosi fattori, tra cui la frammentazione dell’habitat e il conseguente isolamento delle popolazioni, le mutazioni e gli incroci spontanei, il sistema propagativo e il flusso genico, la deriva genetica e la selezione naturale ([21], [20]). Nel caso di specie coltivate o comunque utilizzate dall’uomo, occorre considerare anche l’effetto dei trasferimenti di germoplasma, della selezione artificiale e della diffusione di genotipi ritenuti di maggior interesse. È questo il caso del noce comune (Juglans regia L.), la cui distribuzione è dovuta principalmente all’attività umana, come conseguenza della molteplicità dei suoi impieghi. Il centro di origine della specie sembra essere l’Asia centrale, più precisamente la catena montuosa occidentale dell’Himalaya nel Cachemire, Tajikistan e Kyrgyzstan ([10], [11]). In Europa, pare ormai certo il noce fosse presente fin da prima dell’ultima glaciazione, che ne avrebbe causato la migrazione verso est. Sebbene alcuni esemplari siano sopravvissuti come relitti ([15]), la ricolonizzazione post-glaciale più massiccia si sarebbe verificata circa 7000 anni fa, a partire da materiali provenienti dall’Asia, attraverso l’Iran, la Turchia e i Balcani. Anche il nostro Paese, tuttavia, stando a quanto rilevato da fossili di polline e di frutti, potrebbe essere un centro di origine della specie ([11]).
Attualmente, il noce si trova nel nostro Paese a livello di individui sparsi o in piccoli gruppi, distribuiti lungo i bordi di aree coltivate, lungo le rive dei fiumi o nei centri urbani. Esso è presente in tutte le regioni italiane e si spinge fino a quote di 1.000-1.200 (eccezionalmente 1.500) metri. Le piante sono generalmente isolate e di origine incerta: soltanto in alcuni casi è possibile risalire ad una specifica cultivar o a progenie da questa derivata per libera impollinazione ([11]).
La pressione selettiva esercitata nel tempo dall’uomo per la produzione di frutti di qualità e la contrazione delle aree dedicate alla coltivazione di questa pianta hanno portato alla riduzione delle risorse genetiche disponibili. Questo ha contribuito a ridurre notevolmente la base genetica da impiegare per la selezione ed il miglioramento per caratteri fenotipici utili per la produzione di legname di qualità ([7]). Diventa pertanto sempre più importante predisporre piani per la salvaguardia e la conservazione della biodiversità propria della specie. Una delle fasi in cui è possibile avvengano perdite di variabilità genetica è quella dell’approvvigionamento di materiale propagativo, soprattutto se questo è effettuato senza tenere in considerazione le indicazioni scientifiche oggi disponibili per la sua conservazione ([1]). Inoltre, perdite di variabilità genetica si possono verificare durante tutte le fasi della filiera che dal popolamento di origine portano al materiale da impiegare nei rimboschimenti: i momenti più critici appaiono quelli della raccolta dei semi e della loro lavorazione in stabilimento, nonché le condizioni di allevamento nei vivai ([8]).
Scopo della ricerca è stato l’esame delle caratteristiche genetiche del noce in Piemonte, per valutarne l’entità e la distribuzione della variabilità, e per definire le più opportune strategie di approvvigionamento di seme in tale regione. In particolare, si è inteso verificare se piante che crescono in aree vicine e omogenee dal punto di vista ecologico presentino similitudini genetiche tra di loro o se, al contrario, le eventuali differenze riscontrate siano in qualche modo correlabili alle caratteristiche ecologiche dei siti di impianto nonché alle loro distanze geografiche.
Materiali e Metodi
Materiale vegetale
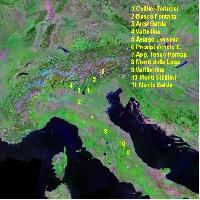
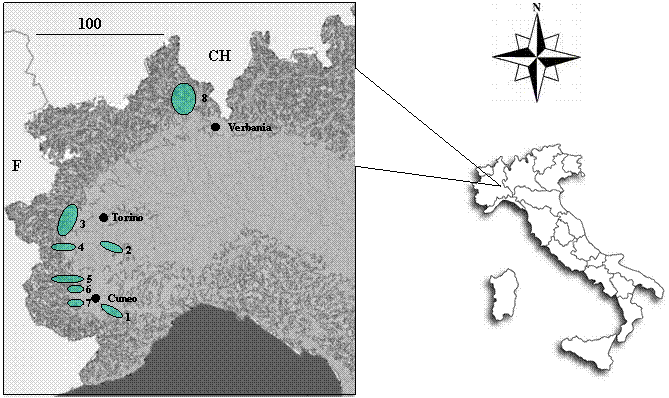
Gli individui oggetto di analisi sono stati scelti nell’ambito di aree segnalate dall’IPLA S.p.A. (Istituto per le Piante da Legno e l’Ambiente, Torino), sulla base della rappresentatività delle diverse situazioni ecologiche in cui il noce è diffuso in Piemonte. La loro localizzazione è riportata in Tab. 1 ed in Fig. 1. L’area denominata “torinese” (n. 3) presenta una superficie superiore rispetto alle altre: ciò è dovuto al ridotto numero di individui presenti al suo interno, che ha sconsigliato una maggior frammentazione dell’area stessa. Si sottolinea che all’interno dell’area considerata non esistono barriere in grado di ostacolare il flusso pollinico tra i vari individui.
Tab. 1 - Localizzazione geografica dei siti oggetto di campionamento.
| N. pianta | Prov. | Area | Località di campionamento (popolazione) | Coordinate geografiche | ||
|---|---|---|---|---|---|---|
| da | a | Lat. N | Long. E | |||
| 1 | 17 | Cuneo | Valle Pesio | Chiusa Pesio, F.ne Comba | 44° 21’ | 7° 41’ |
| Peveragno, Montefallonio | 44° 19’ | 7° 39’ | ||||
| Boves, S. Antonio | 44° 19’ | 7° 32’ | ||||
| Boves, Pasturone | 44° 20’ | 7° 32’ | ||||
| Boves, Finetti-Balotin | 44° 19’ | 7° 32’ | ||||
| 18 | 27 | Cuneo | Pianura | Monasterolo, C.na Riforano | 44° 41’ | 7° 36’ |
| Villanova Solaro, M.na Noce | 44° 43’ | 7° 35’ | ||||
| 28 | 31 | Torino | Torinese | Giaveno, Sala | 45° 06’ | 7° 21’ |
| 47 | 51 | Giaveno, Villa | 45° 02’ | 7° 22’ | ||
| - | - | Bobbio Pellice, C. Carboneri | 44° 48’ | 7° 08’ | ||
| 32 | 46 | Cuneo | Valle Po | Paesana, Seymandi | 44° 40’ | 7° 17’ |
| Robella, S. Chiaffredo | 44° 40’ | 7° 18’ | ||||
| Rifreddo, Monastero | 44° 39’ | 7° 21’ | ||||
| 52 | 66 | Cuneo | Val Maira | Cartignano, Cimitero | 44° 29’ | 7° 17’ |
| Cartignano, Tetti | 44° 26° | 7° 18’ | ||||
| Dronero, Pratavecchia | 44° 27’ | 7° 24’ | ||||
| 67 | 76 | Cuneo | Val Grana | Monterosso, Valle | 44° 25’ | 7° 20’ |
| Caraglio, Monte | 44° 25’ | 7° 23’ | ||||
| 77 | 86 | Cuneo | Valle Stura | Roccasparvera, M.na Grazie | 44° 21’ | 7° 26’ |
| Moiola, Valle | 44° 19’ | 7° 24’ | ||||
| 87 | 104 | Verbania | Ossola | Crodo, Smeglio | 46° 13’ | 8° 19’ |
| Baceno, P.te Silogno | 46° 15’ | 8° 19’ | ||||
| Premia, Bivio Uderzo | 46° 16’ | 8° 21’ | ||||
| Pioda Bivio Cresta | 46° 16’ | 8° 20’ | ||||
Fig. 1 - Localizzazione delle aree geografiche oggetto di campionamento. 1 = Valle Pesio, 2 = Pianura cuneese, 3 = Torinese, 4 = Valle Po, 5 = Valle Maira, 6 = Valle Grana, 7 = Valle Stura, 8 = Ossola.
I campioni erano costituiti sia da piante isolate che da soggetti presenti in popolazioni più o meno ampie: in questo caso l’analisi ha coinvolto fino a 5 individui per popolazione, scelti con criteri di casualità. Tutte le piante campionate erano adulte, con un’età stimata non inferiore a 30 anni. Non è stato possibile risalire con sufficiente certezza all’origine del materiale da cui le piante sono derivate, anche se si può ipotizzare, con ragionevole certezza, che si tratti di piante nate da seme locale, non innestate e non appartenenti a particolari cultivar o varietà.
Il campionamento ha previsto il prelievo di gemme invernali, raccolte durante il riposo vegetativo delle piante. Se l’estrazione del DNA non è avvenuta immediatamente dopo la raccolta, il materiale è stato conservato, fino al momento dell’utilizzazione, a - 20°C.
Analisi di laboratorio
La stima della variabilità genetica è stata effettuata impiegando marcatori RAPD (Random Amplifed Polimorphic DNA). Il DNA, estratto con metodo CTAB ([6]) leggermente modificato, è stato successivamente amplificato con procedimento PCR (Polymerase Chain Reaction).
Per l’amplificazione sono stati utilizzati i primers indicati in Tab. 2: si tratta di primers decamerici, prescelti sulla base del polimorfismo evidenziato nell’ambito di ricerche precedenti ([11]). La miscela di reazione era composta da buffer di reazione 1x, MgCl2 2.5 M, dNTP mix 0.2 μM, primer 0.2 μM, 1 unità Taq Polimerasi (Promega), DNA campione 20 ng e H2 O bidistillata sterile necessaria per raggiungere un volume finale di 25 μl. La procedura PCR ha seguito un protocollo comprendente una fase iniziale di 5 minuti a 95°C, 40 cicli (composti da una fase di denaturazione di 1 minuto a 94°C, una fase di annealing di 1 minuto a 36°C e una fase di sintesi di 2 minuti a 72°C) e una fase finale di 8 minuti a 72°C. La separazione elettroforetica delle bande ottenute dall’amplificazione è stata effettuata su gel di agarosio all’1.5%, tenuto a 65 V per 4 ore. L’intero procedimento è stato ripetuto due volte per ciascun primer al fine di verificare la ripetibilità delle analisi: nel caso di mancata conferma dei risultati, le bande non sono state considerate. Nel gel è stato incorporato bromuro di etidio, che ha permesso la visualizzazione del DNA agli UV, ponendo il gel a fine corsa in un fluorimetro associato ad un digitalizzatore di immagini (GelDoc). I pattern elettroforetici così evidenziati sono stati successivamente elaborati per l’analisi statistica.
Tab. 2 - Primers RAPD decamerici utilizzati per l’amplificazione del DNA.
| Primer | Sequenza nucleotidica (da 5’ a 3’) | Rapporto di basi GC/AT (%) |
|---|---|---|
| OPA02 | TGCCGAGCTG | 70 |
| OPA09 | GGGTAACGCC | 70 |
| OPA12 | TCGGCGATAG | 60 |
| OPA18 | AGGTGACCGT | 60 |
| OPE04 | GTGACATGCC | 60 |
| OPE07 | AGATGCAGCC | 60 |
| OPE15 | ACGCACAACC | 60 |
| OPE18 | GGACTGCAGA | 60 |
| OPE19 | ACGGCGTATG | 60 |
| OPE20 | AACGGTGACC | 60 |
Elaborazione dei dati
La stima dei parametri genetici della popolazione di piante considerata è stata calcolata usando il programma statistico POPGENE 1.21 ([24]). La variabilità genetica e la ricchezza allelica sono state stimate ricorrendo al computo della diversità genetica (He), del numero medio (N) e del numero effettivo (Ne) di alleli per locus e della percentuale di loci polimorfici (P) ([14], [5]). Le distanze genetiche tra singoli individui sono state misurate ricorrendo al parametro ΦST ([23]). Tali valori sono stati utilizzati per elaborare il dendrogramma delle distanze genetiche secondo il metodo UPGMA (Unweighted Pair-Group Method using Arithmetic means - [22]) ed utilizzando il programma NTSYS-pc 2.10j ([19]). La ripartizione della diversità genetica è stata effettuata attraverso l’analisi della varianza molecolare (AMOVA), utilizzando il software GENALEX 6 ([16]): in particolare, la diversità genetica è stata valutata tra aree geografiche e, all’interno di queste, tra popolazioni, costituite da piante campionate nella stessa località. Lo stesso software è stato utilizzato per valutare le relazioni tra aree geografiche mediante l’analisi delle Coordinate Principali (PCoA), ricavate dalle distanze genetiche e proiettate in un grafico bidimensionale.
Il Mantel test ([12]) è stato utilizzato per correlare le matrici delle distanze genetiche e di quelle geografiche, valutate come la distanza in linea d’aria che intercorre tra ciascuna coppia di popolazioni. Il risultato è stato validato ricorrendo a 9999 permutazioni, utilizzando il programma GENALEX 6.
Risultati
L’amplificazione del DNA mediante l’uso dei 10 primers prescelti ha consentito l’identificazione di 101 bande. Ciascuna di esse è stata indicata con la sigla del primer corrispondente, seguita dal peso molecolare del frammento, espresso in numero di coppie di basi azotate (bp). I marcatori RAPD, valutando la presenza ( oppure 1) o l’assenza (- oppure 0) della banda, identificano l’esistenza o meno di una sequenza nucleotidica (quella complementare al primer, che, se presente, consente il processo di amplificazione del DNA e, quindi, la comparsa della banda). Le caratteristiche generali delle bande ottenute nell’ambito della prova di caratterizzazione delle piante sono riportate in Tab. 3.
Tab. 3 - Caratteristiche delle bande ottenute a seguito di amplificazione del DNA.
| Primer | N. totale frammenti | Peso mol. (range in bp) | Frammenti polimorfici | Frammenti monomorfici | ||
|---|---|---|---|---|---|---|
| Numero | Peso mol. (bp) | Numero | Peso mol. (bp) | |||
| OPA02 | 13 | 340-1120 | 7 (54%) | 440, 580, 640, 670, 820, 880, 1020 | 6 (46%) | 340, 520, 700, 750, 960, 1120 |
| OPA09 | 12 | 340-1260 | 6 (50%) | 400, 540, 680, 860, 1150, 1260 | 6 (50%) | 340, 510, 640, 720, 800, 1050 |
| OPA12 | 7 | 330-1760 | 3 (43%) | 960, 1090, 1160 | 4 (57%) | 330, 620, 1260, 1760 |
| OPA18 | 13 | 380-1420 | 4 (31%) | 680, 800, 900, 960 | 9 (69%) | 380, 500, 530, 600, 750, 860, 1160, 1280, 1420 |
| OPE04 | 9 | 590-1050 | 2 (22%) | 880, 950 | 7 (78%) | 590, 740, 1050, 1100, 1170, 1220, 1400 |
| OPE07 | 6 | 460-1350 | 3 (50%) | 460, 530, 810 | 3 (50%) | 950, 1120, 1350 |
| OPE15 | 11 | 600-1200 | 5 (45%) | 770, 840, 950, 1020, 1050 | 6 (55%) | 600, 650, 700, 980, 1160, 1200 |
| OPE18 | 9 | 670-1270 | 5 (56%) | 670, 820, 850, 950, 1230 | 4 (44%) | 710, 1010, 1100, 1270 |
| OPE19 | 9 | 530-1600 | 5 (56%) | 530, 700, 760, 1460, 1600 | 4 (44%) | 580, 630, 860, 1160 |
| OPE20 | 12 | 540-1400 | 8 (67%) | 540, 570, 650, 750, 970, 1000, 1070, 1370 | 4 (33%) | 780, 870, 1100, 1400 |
| Totale | 101 | - | 48 (47.5%) | - | 53 (52.5%) | - |
Dall’esame dei dati si può osservare che il numero medio di bande per primer è pari a 10.1, con peso molecolare variabile da 330 a 1.760 bp. Cinquantatre bande su 101 (poco più del 50%) sono risultate monomorfiche, cioè presenti in tutti i campioni, i quali pertanto risultano caratterizzati da un solo tipo di allele.
La Tab. 4 riporta i valori di ricchezza allelica e di diversità genetica (assumendo come valido l’equilibrio di Hardy-Weinberg). Il numero medio di alleli per locus è risultato variare tra 1.30 (Val Grana) e 1.39 (Ossola), con valore medio di 1.36; analogamente il numero effettivo di alleli per locus è variato da 1.20 (Val Grana) a 1.26 (Valle Pesio), con valore medio di 1.23. La percentuale di loci polimorfici ha confermato l’andamento generale: valore medio di 35.2 con estremi di variazione compresi tra 29.7 (Val Grana) e 38.6 (Ossola); valori elevati sono stati riscontrati anche nelle valli Pesio e Maira (rispettivamente 36.7 e 37.6). La diversità genetica, infine, è variata da 0.114 (Val Grana) a 0.143 (Ossola), con valore medio di 0.131 e risultati sensibilmente superiori alla media registrati anche in Valle Pesio.
Tab. 4 - Valori di ricchezza allelica e di diversità genetica nell’ambito delle aree geografiche oggetto di analisi.
| Popolamento | Numero medio alleli per locus (N) | Numero effettivo alleli per locus (Ne) | Percentuale loci polimorfici (P) | Diversità genetica (He) |
|---|---|---|---|---|
| 1. Valle Pesio | 1.37 | 1.26 | 36.7 | 0.141 |
| 2. Pianura | 1.33 | 1.22 | 32.7 | 0.123 |
| 3. Torinese | 1.36 | 1.24 | 35.6 | 0.137 |
| 4. Valle Po | 1.36 | 1.23 | 35.6 | 0.132 |
| 5. Val Maira | 1.38 | 1.23 | 37.6 | 0.133 |
| 6. Val Grana | 1.30 | 1.20 | 29.7 | 0.114 |
| 7. Valle Stura | 1.35 | 1.23 | 34.7 | 0.128 |
| 8. Ossola | 1.39 | 1.25 | 38.6 | 0.143 |
| Media | 1.36 | 1.23 | 35.2 | 0.131 |
| Dev. standard | 0.03 | 0.02 | 2.7 | 0.009 |
Per quanto concerne i livelli di differenziazione genetica sono state condotte due elaborazioni distinte: tra tutti i singoli individui e tra aree geografiche. Nella prima, come si può osservare dal dendrogramma riportato in Fig. 2, soltanto in pochi casi è stato possibile evidenziare similitudini genetiche tra individui appartenenti alla stessa area geografica ed addirittura alla stessa popolazione (ad esempio per le piante n. 24, 25 e 26 raccolte a Villanova Solaro oppure per quelle n. 43, 44 e 46, provenienti dalla popolazione di Monastero a Rifreddo).
Fig. 2 - Cluster analisi ottenuta dalle distanze genetiche (ΦST) tra piante singole mediante metodo UPGMA.

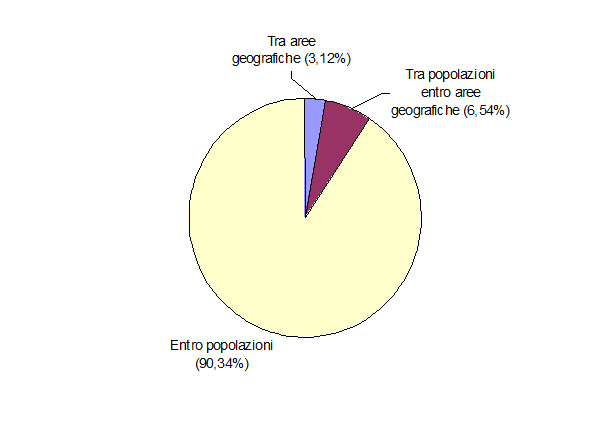
La differenziazione tra aree geografiche, stimata mediante l’analisi AMOVA (Tab. 5), è responsabile del 3.12% della variabilità genetica totale, mentre la diversità tra popolazioni appartenenti alla stessa area è pari al 6.54% della varianza totale (Fig. 3). Come previsto, la più alta percentuale di variabilità genetica (90.34%) è stata riscontrata entro le popolazioni.
Tab. 5 - Analisi della varianza molecolare (AMOVA) tra aree geografiche e popolazioni oggetto di analisi.
| Fonte di variazione | Gradi di libertà | Somma dei quadrati | Componenti della varianza | Percentuale di variazione | P |
|---|---|---|---|---|---|
| Tra aree geografiche | 7 | 78757 | 11251 | 3.12 | < 0.01 |
| Tra popolazioni | 14 | 117190 | 8371 | 6.54 | < 0.01 |
| Entro popolazioni | 82 | 513533 | 6263 | 90.34 | < 0.01 |
| TOTALE | 103 | 709481 | 25884 | 100 | - |
Fig. 3 - Ripartizione della varianza molecolare nelle sue componenti tra popolazioni, tra aree geografiche e tra popolazioni entro area geografica.
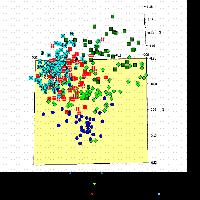
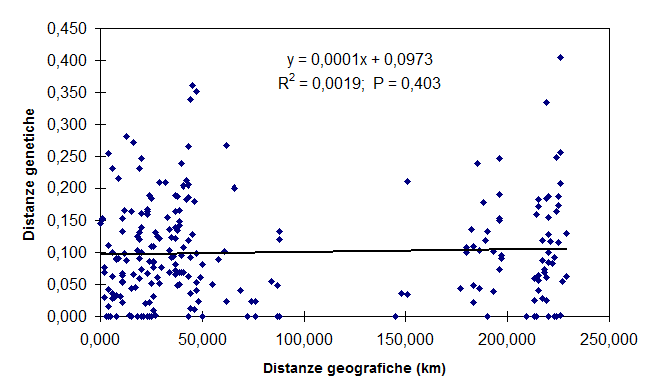
L’analisi delle coordinate principali ha consentito di individuare due set di variabili che spiegano, rispettivamente, il 59.63 e il 20.43% della variabilità complessiva. Dall’analisi del relativo grafico (Fig. 4) si può osservare che la disposizione delle aree nel plot non rispetta la loro localizzazione geografica. Tale dato è confermato dal risultato del test di Mantel (Fig. 5), il quale non evidenzia alcuna relazione statisticamente significativa tra distanze genetiche e distanze geografiche delle singole popolazioni (P = 0.403).
Fig. 4 - Analisi delle coordinate principali (PCoA) ricavata dalla matrice delle distanze genetiche tra le aree geografiche analizzate. La prima coordinata spiega il 59.63% della variabilità complessiva, la seconda il 20.43%.
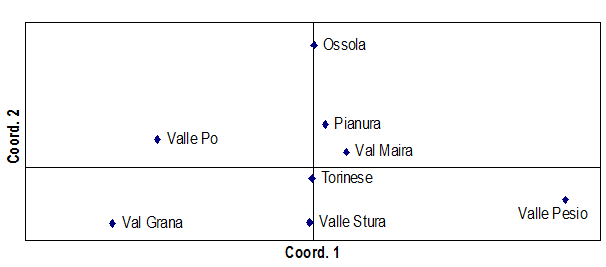
Fig. 5 - Correlazione tra distanze genetiche (ΦST) e distanze geografiche tra le popolazioni esaminate (Mantel test). Non si evidenzia alcuna relazione statisticamente significativa tra i due parametri (P = 0.403).
Discussione
I risultati della ricerca hanno confermato come anche in Piemonte le caratteristiche genetiche delle popolazioni di noce non siano dissimili da quelle evidenziate in altre aree italiane ([11], [17], [18]). Il materiale ha presentato un elevato grado di uniformità genetica, comprovato dal fatto che circa il 50% dei marcatori RAPD è risultato monomorfico. A titolo di confronto si riportano i risultati di analoghe ricerche, condotte in aree geograficamente paragonabili a quella oggetto del presente studio, ma su altre specie: nel ciavardello la frequenza di bande RAPD monomorfiche è risultata limitata al 17% ([2]), mentre nella farnia solo 4 marcatori su un totale di 104 sono risultati monomorfici e non hanno quindi presentato variabilità ([4]). Un elevato livello di uniformità genetica nell’ambito del materiale oggetto del presente studio è stato evidenziato anche dall’analisi di marcatori microsatelliti nucleari (dati non riportati): delle quattro coppie di primer analizzate, infatti, solamente due hanno presentato polimorfismo, e per di più ad un livello molto modesto (rispettivamente due e tre alleli). A causa di tale ridotto polimorfismo i dati relativi a questo tipo di marcatori (che, essendo codominanti, avrebbero permesso di distinguere gli eterozigoti dagli omozigoti) non sono stati considerati per la presente ricerca. Anche in questo caso, il contrasto con i valori ricavati dalle analisi condotte su altre specie appare stridente: nel frassino maggiore sono stati individuati 253 alleli in 6 loci microsatelliti ([9]), mentre nel pino silvestre 205 alleli in 7 loci ([3]). È tuttavia opportuno far notare come, in queste ultime due ricerche, l’areale sottoposto ad indagine fosse di maggiori dimensioni, essendo esteso a tutta l’Italia settentrionale.
Da quanto detto è lecito pensare che la scarsa variabilità genetica del noce comune nel nostro Paese sia per lo più una diretta conseguenza delle attività umane. La specie è, infatti, una delle piante arboree più legate alla storia dell’uomo ed è noto come la domesticazione sia causa di profondi cambiamenti nelle caratteristiche genetiche di una specie, soprattutto a causa della selezione artificiale che finisce con l’erodere il pool genetico naturale ([13]). La diffusione di poche piante o varietà dalle caratteristiche di pregio va a discapito degli individui appartenenti a popolazioni più antiche, con evidenti conseguenze di erosione genetica. La riduzione della diversità e la potenziale perdita di risorse genetiche per la specie sono indicate anche dall’assenza di un chiaro pattern geografico della variabilità genetica evidenziata.
Una delle possibili fonti di variazione potrebbe essere collegata alla quota altimetrica dei siti ove si trovano le popolazioni, la quale potrebbe aver favorito la diffusione artificiale di materiale genetico con specifiche caratteristiche di adattamento. Tuttavia, i dati della presente ricerca non hanno evidenziato significative differenze tra popolazioni di pianura e popolazioni di fondo valle, probabilmente anche a causa dell’esiguo range di variazione altimetrica considerato (da 270 m s.l.m. della pianura cuneese ai 750 dell’Ossola).
Da questa elevata omogeneità genetica discende una scarsa differenziazione dei gruppi di piante esaminati, che risultano essere distribuiti sul territorio in un modo che riflette presumibilmente la modalità di approvvigionamento del materiale utilizzato per la propagazione degli individui. Popolazioni geograficamente vicine o che crescono in condizioni ecologiche simili risultano solo in pochi casi geneticamente più simili tra loro rispetto a popolazioni cresciute in condizioni diverse. Analogamente, costituiscono un’eccezione i casi in cui individui della stessa popolazione sono geneticamente più simili rispetto a piante che crescono in località differenti.
Di conseguenza, l’approvvigionamento di materiale di propagazione da queste piante potrà essere effettuato seguendo come criterio soprattutto il valore fenotipico degli individui, il quale risulta strettamente legato all’adattamento alle particolari condizioni ambientali del luogo in cui la pianta cresce. Pertanto, per una specie come il noce, propagata quasi del tutto artificialmente, applicare il metodo delle Regioni di Provenienza (così come previsto dalla vigente normativa a livello comunitario e nazionale) assume una valenza sfumata. Tuttavia, non è possibile escludere che ricorrendo a marcatori caratterizzati da un maggiore livello di polimorfismo (ad esempio ISSR) oppure ampliando il numero di marcatori RAPD analizzati, sia possibile evidenziare maggiori livelli di diversità genetica, che potrebbero consentire una più dettagliata verifica delle differenze genetiche. L’uso congiunto di diversi tipi di marcatori (morfologici, fenologici, biochimici e molecolari) potrebbe essere il modo migliore per caratterizzare le provenienze.
Ringraziamenti
Ricerca svolta nell’ambito del Progetto RI.SELV.ITALIA (Sottoprogetto 1.1: Biodiversità e produzione di materiale forestale di propagazione) e con il sostegno finanziario della Regione Piemonte, Direzione Economia Montana e Foreste.
References
Google Scholar
Google Scholar
Google Scholar
Google Scholar
Google Scholar
Google Scholar
Google Scholar
Google Scholar
CrossRef | Google Scholar
Google Scholar
Google Scholar
Google Scholar
Google Scholar
CrossRef | Google Scholar
Google Scholar
CrossRef | Google Scholar
Google Scholar
CrossRef | Google Scholar
Google Scholar
CrossRef | Google Scholar
CrossRef | Google Scholar
Google Scholar
CrossRef | Google Scholar
Online | Google Scholar